What if my data comes from a long-read sequencing technology…
When working with long-read sequencing (LRS), you will typically obtain full-length transcripts with IDs that are specific to your experiment, and therefore are not supported by tappAS. Still, you can use tappAS to study your LRS dataset but additional steps are required.
To make LRS data compatible with tappAS, we make use of the SQANTI3 and IsoAnnotLite tools.
(The current version of IsoAnnotLite is 2.7.3, make sure you use the last version of the script).
SQANTI3 is a pipeline for the structural characterization of isoforms obtained by full-length transcript sequencing. SQANTI3 takes full-length transcript sequences in FASTA format, which can be obtained after Iso-seq3 (PacBio) or FLAIR (Nanopore) processing. The only additional requirement is that the species genome and transcriptome files are available, therefore SQANTI3 is restricted today to work with sequenced species. SQANTI3 provides a wide range of descriptors of transcript quality and generates a graphical report to aid in the interpretation of LRS results. More information on SQANTI3 can be obtained here.
There are two ways to transform your SQANTI3 output to tappAS:
- Transform structural information. In this scenario, the SQANTI3 transcript types (FSM, ISM, NIC, etc) are processed into a tappAS gff3 file containing this information. This can be fed to tappAS to visualize transcript models and study differential expression, isoform usage, and UTR analysis. However, no functional analysis is possible here.
- Transform structural information and add functional data. This is the recommended option if your species is supported by tappAS. In this case, you make use of the tappAS species specific GFF3 to map functional elements to your LRS dataset. Please, bear in mind that in this case, you must run SQANTI3 with the same reference genome annotation as used in tappAS.
IsoAnnotLite is a python3 script that takes a SQANTI3 output, and optionally a tappAS precomputed gff3 file, and returns a new gff3 file fully compatible with the tappAS software.
How to proceed
There are four basic steps you need to follow:
As an example, the next command will be a form to call the script with a gff3 reference to use as annotation:
- Use SQANTI with your LRS fasta file to obtain «_corrected.gtf», «_classification.txt» and «_junctions.txt» files.
- If your species is supported by tappAS, download the corresponding GFF3 here.
- Download the IsoAnnotLite, script and unzip.
- Run the basic IsoAnnotLite command as indicated below (note that all arguments are optional [use -h for more information]).
python3 IsoAnnotLite.py my_corrected.gtf my_classification.txt my_junctions.txt -gff3 tappAS.gff3 -o output_name_newGFF3 -stdout output_name_statisticalResults -novel -nointronic -saveTranscriptIDs
Argument -novel allows IsoAnnotLite to compare every transcript against all the transcripts that belong to the same gene. This procedure takes more time but gets more annotations. However, it is not recommended for those transcripts that already have a reference in the GFF3 file.
If you do not use it, IsoAnnotLite will use this method only for those transcripts that do have not a reference transcript in the SQANTI output or do not have features information to get from the reference transcript (novel transcripts).
Example of use:
python3 IsoAnnotLite.py PacBio_corrected.gtf PacBio_classification.txt PacBio_junctions.txt -gff3 Mus_musculus_Ensembl_86.gff3
Note that SQANTI3 files should be provided in the order indicated above. In case of problems, use the argument «-h» to get help.
How were did we generate the GFF3 files?
Precomputed GFF3s were produced following the IsoAnnot pipeline. IsoAnnot requires three pieces of information as input data: isoform sequences and ORF sequences associated with them in fasta format, and gene models in GTF format. If the user is annotating reference transcriptomes, all of these files are downloaded from Ensembl or RefSeq databases. If the user wants to annotate a long-reads defined transcriptome, they only need to provide the isoform sequences in FASTA format.
First of all, IsoAnnot uses SQANTI (1) to generate a structural annotation and classification by comparing the isoforms splice junctions to the reference transcriptome. When the input is a long-reads defined transcriptome, SQANTI also outputs the predicted protein sequences and the GTF file necessary to proceed with the functional annotation.
The IsoAnnot pipeline comprises several modules that integrate coordinate-defined functional annotations at RNA and protein level derived from public databases and sequence-based prediction tools:
- RNA-level annotations include cis-acting UTR regulatory elements, upstream open reading frames (uORFs) and polyadenylation sites predicted by UTRscan (2) and repeat regions and low-complexity elements predicted by RepeatMasker (3,4). Also, isoforms containing a premature termination codon (PTC)—potentially leading to nonsense-mediated decay (NMD)— are predicted using the 50-nt rule(5) that indicates that a termination codon situated more than 50–55 nt upstream of an exon-exon junction is generally a PTC. Additionally, annotation of miRNA binding sites relies on the mirWalk2.0 (6) and miRBase (7) databases. miRNAs and its interactions are filtered based on the experimental evidence level and the number of sources reporting the associations. Then, as miRNA-mRNA interaction sites are provided by mirWalk2.0 in transcript coordinates, we map them to genomic coordinates. Finally, this genome-coordinate annotation is used to transfer the miRNA binding sites to query splicing variants. Only complete, contiguous and strand-specific matches of the seed region in the query isoform are annotated by IsoAnnot.
- At protein level, Pfam domains (8) are mapped with InterProScan5 (9), transmembrane regions and nuclear localization signals (NLS) are predicted with TMHMM (10) and NucImport(11), and signal peptides coiled-coil and disordered regions are obtained from SignalP 4.1 (12), COILS (13) and MobiDB Lite (14,15) respectively. In addition, information on protein functional features is retrieved by parsing UniProtKB (16) and PhosphoSitePlus (17) databases. This is done by using the cross-reference information between UniProt and Ensembl and RefSeq databases to retrieve the genomic coordinates of the features to be annotated. Then, these features are transferred to the query isoforms and are kept in the final annotation if the original sequence matches the one present in the query isoform.
We complemented functional information at the isoform-resolution level with labels that describe the processes and pathways in which those genes are involved, considering data from GO (18,19) and Reactome (20) .
IsoAnnot produces a GFF3-like file with the structural and functional annotation of the transcriptome which can be read by tappAS (21) to perform further statistical analyses.
The pipeline has been used to annotate reference transcriptomes of model organisms from Ensembl and RefSeq databases. IsoAnnot provides substantial functional information at the isoform-resolution for well studied model organisms. At the protein level, more than 90% of coding isoforms were annotated with at least one functional feature for both databases and organisms (Table 1). However, differences inherent in the definition, nature and proportion of non-coding isoforms between alternative reference sources (Table 1) led to higher differences in the global annotation coverage reached by IsoAnnot (70% vs 90% of isoforms with at least one annotated element for Ensembl and RefSeq, respectively (Table 1).
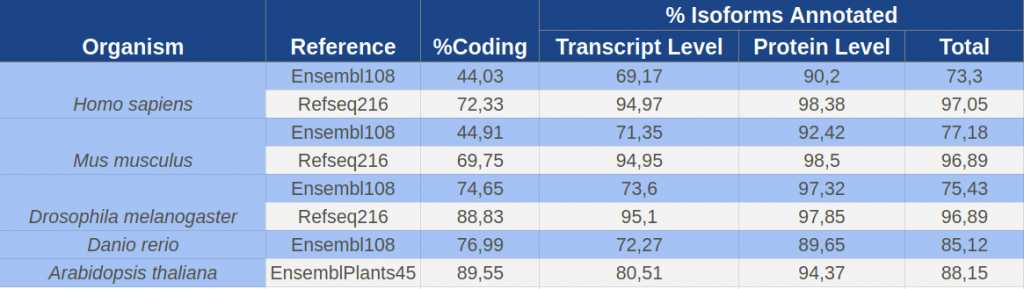
Table 1. Number of transcripts, proportion of coding transcripts and proportion of isoforms annotated at the transcript and protein levels for the different reference transcriptomes annotated by IsoAnnot pipeline.
References
1. Tardaguila M, De La Fuente L, Marti C, Pereira C, Pardo-Palacios FJ, Del Risco H, et al. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 2018 Mar;28(3):396–411.
2. Grillo G, Turi A, Licciulli F, Mignone F, Liuni S, Banfi S, et al. UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010 Jan;38:D75–80.
3. Using and Understanding RepeatMasker. In: Mobile Genetic Elements: Protocols and Genomic Applications. Totowa, NJ: Humana Press; 2012. p. 29–51. (Methods in Molecular Biology; vol. 859).
4. Smit, AFA, Hubley, R & Green. P. RepeatMasker Open-4.0. http://www.repeatmasker.org; 2013.
5. Zhang Z, Xin D, Wang P, Zhou L, Hu L, Kong X, et al. Noisy splicing, more than expression regulation, explains why some exons are subject to nonsense-mediated mRNA decay. BMC Biol. 2009 May 14;7(1):23.
6. Sticht C, De La Torre C, Parveen A, Gretz N. miRWalk: An online resource for prediction of microRNA binding sites. PLOS ONE. 2018 Oct 18;13(10):e0206239.
7. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019 Jan 8;47(D1):D155–62.
8. Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021 Jan 8;49(D1):D412–9.
9. Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014 May 1;30(9):1236–40.
10. Krogh A, Larsson B, Von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes11Edited by F. Cohen. J Mol Biol. 2001 Jan;305(3):567–80.
11. Mehdi AM, Sehgal MSB, Kobe B, Bailey TL, Bodén M. A probabilistic model of nuclear import of proteins. Bioinformatics. 2011 May 1;27(9):1239–46.
12. Predicting Secretory Proteins with SignalP. In: Protein Function Prediction: Methods and Protocols. Springer; 2017. p. 59–73. (Methods in Molecular Biology; vol. 1611).
13. Lupas A, Van Dyke M, Stock J. Predicting Coiled Coils from Protein Sequences. Science. 1991 May 24;252(5009):1162–4.
14. Necci M, Piovesan D, Dosztányi Z, Tosatto SCE. MobiDB-lite: fast and highly specific consensus prediction of intrinsic disorder in proteins. Bioinformatics. 2017 May 1;33(9):1402–4.
15. Potenza E, Domenico TD, Walsh I, Tosatto SCE. MobiDB 2.0: an improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res. 2015 Jan 28;43(D1):D315–20.
16. The UniProt Consortium, Bateman A, Martin MJ, Orchard S, Magrane M, Ahmad S, et al. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D523–31.
17. Hornbeck PV, Kornhauser JM, Latham V, Murray B, Nandhikonda V, Nord A, et al. 15 years of PhosphoSitePlus®: integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019 Jan 8;47(D1):D433–41.
18. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000 May;25(1):25–9.
19. The Gene Ontology Consortium, Aleksander SA, Balhoff J, Carbon S, Cherry JM, Drabkin HJ, et al. The Gene Ontology knowledgebase in 2023. Genetics. 2023 May 2;224(1):iyad031.
20. Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2021 Nov 12;50(D1):D687–92.21. De La Fuente L, Arzalluz-Luque Á, Tardáguila M, Del Risco H, Martí C, Tarazona S, et al. tappAS: a comprehensive computational framework for the analysis of the functional impact of differential splicing. Genome Biol. 2020 Dec;21(1):119.